Esta publicação também está disponível em:
 Português
Português  Español
Español
La amiloidosis cardíaca es cada vez más diagnostica en la práctica clínica. En este texto resumiremos 10 consejos que no puede dejar de conocer sobre esta enfermedad.
- La amiloidosis cardíacaes caracterizada por la depósito extracelular de proteínas anormales (fibrillas de amiloide) en el miocardio, pero también puede afectar a otras estructuras cardiacas tales como la válvula aórtica, las aurículas y sistema de conducción. La simple presencia de amiloides ya aumenta el riesgo de tromboembolismo, incluso a ritmo sinusal.
- En individuos mayores de 80 años, se estima que la amiloidosis cardíaca es más prevalente que la estenosis aórtica (25% vs 10%) y puede exceder el 60% de los pacientes geriátricos. De hecho, la coexistencia entre la estenosis aórtica y la amiloidosis cardíaca es bastante común (entre 4 – 29% de los casos).
- Las dos fibrillas más involucradas en la amiloidosis cardíaca son la transtiretina(TTR, que puede ser mutante [hereditaria] o senil [salvaje]) y la inmunoglobulina de cadena ligera (AL). Por lo tanto, hay 3 tipos de amiloidosis cardíaca : TTR salvaje, TTR hereditario y AL . La TTR es una variante de prealbúmina humana, presente en plasma y LCR. La amiloidosis cardíaca TTR salvaje es la más frecuente de las 3, especialmente en los adultos mayores, y se produce debido a anomalías en el desarrollo y la deposición de TTR, sin involucrar mutación . La amiloidosis TTR hereditaria , por otro lado, es una enfermedad genética con patrón autosómico dominante, observándose en pacientes más jóvenes que en aquellos con TTR salvaje. Entre las 120 mutaciones identificadas en el TTR hereditario, algunas variantes son más comunes, como Val122Ile en la población afroamericana y Val30Met en ciertas regiones de Portugal, Japón y Suecia (comúnmente asociadas con neuropatía).
Repitiendo:
De los tipos de amiloidosis cardíaca, el más común es la transtiretina salvaje o senil.
- La amiloidosis AL, también llamado amiloidosis primaria es la forma más común de la amiloidosis sistémica , y se asocia con un defecto en la producción de inmunoglobulina por células plasmáticas . Puede ocurrir espontáneamente, pero generalmente se asocia con otros trastornos sanguíneos, como el mieloma múltiple y la macroglobulinemia de Waldenström.
- En la amiloidosis cardíaca, la infiltración amiloide comienza a partir de la base em dirección al ápice, resultando en un aumento de espesor y la rigidez del ventrículo, causando miocardiopatía restrictiva / hipertróficacon disfunción diastólica importante y eventual disfunción sistólica asociada:
- La fracción de eyección puede conservarse en las primeras etapas de la amiloidosis cardíaca, pero el 30-50% de los pacientes tendrán algún grado de disfunción sistólica (especialmente en el TTR hereditario).
- Señales de alertapara pensar en la amiloidosis cardíaca:
- Antecedentes de síndrome del túnel carpiano, estenosis del canal lumbar o rotura espontánea del tendón del bíceps(por afectación de tejidos blandos como músculos, tendones y ligamentos). La neuropatía es más común en la forma hereditaria, pero puede afectar al 10% de los pacientes con forma salvaje.
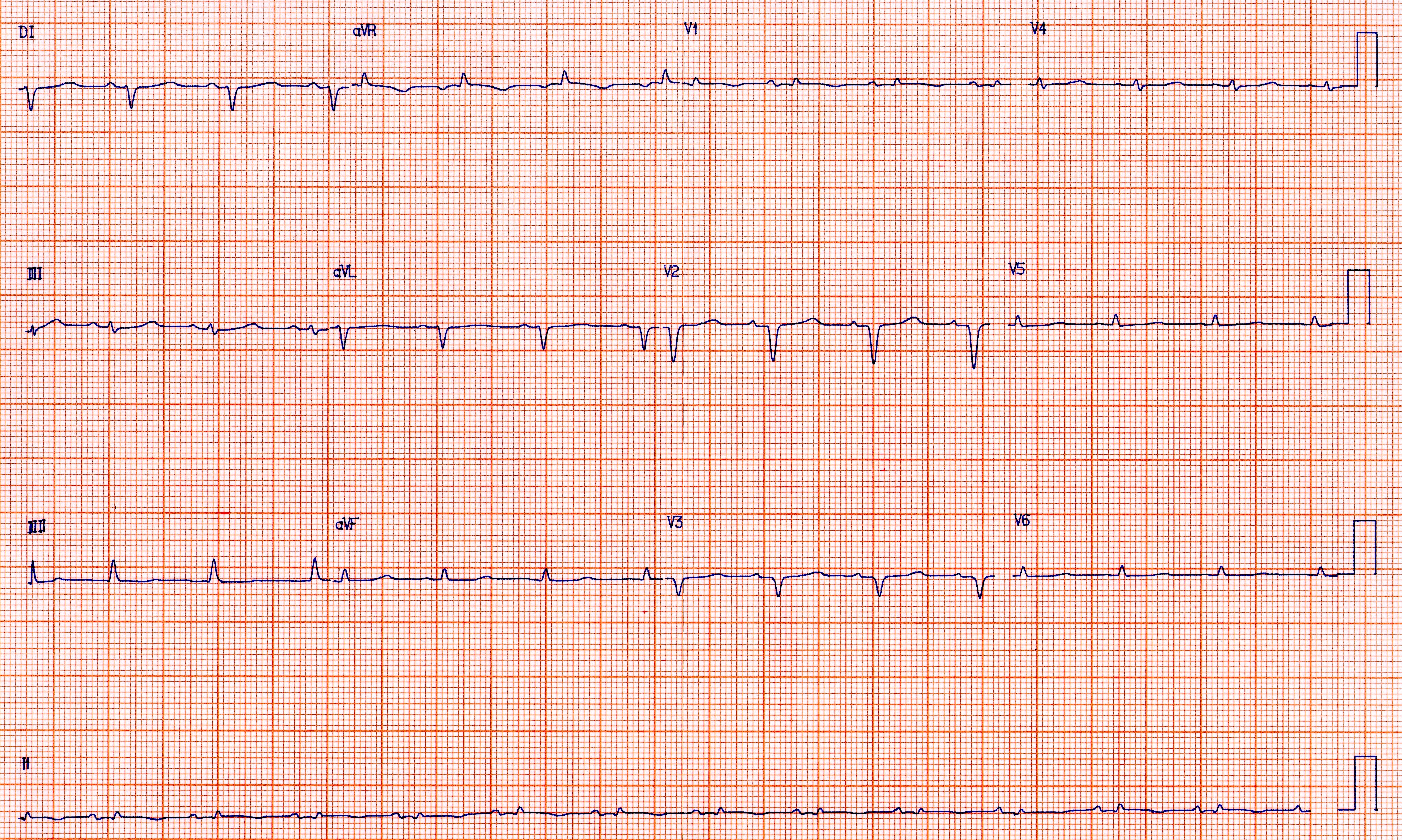
- ECG de bajo voltaje a pesar de la evidencia ecocardiográfica de aumento del grosor ventriculary la presencia de ondas Q sin antecedentes de infarto de miocardio.
- ECG de nuestro banco de imágenes que muestra zonas de bajo voltaje y pseudoinfarto en un paciente con amiloidosis cardíaca.
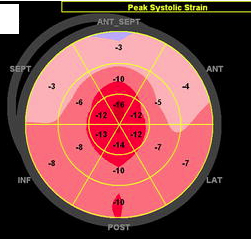
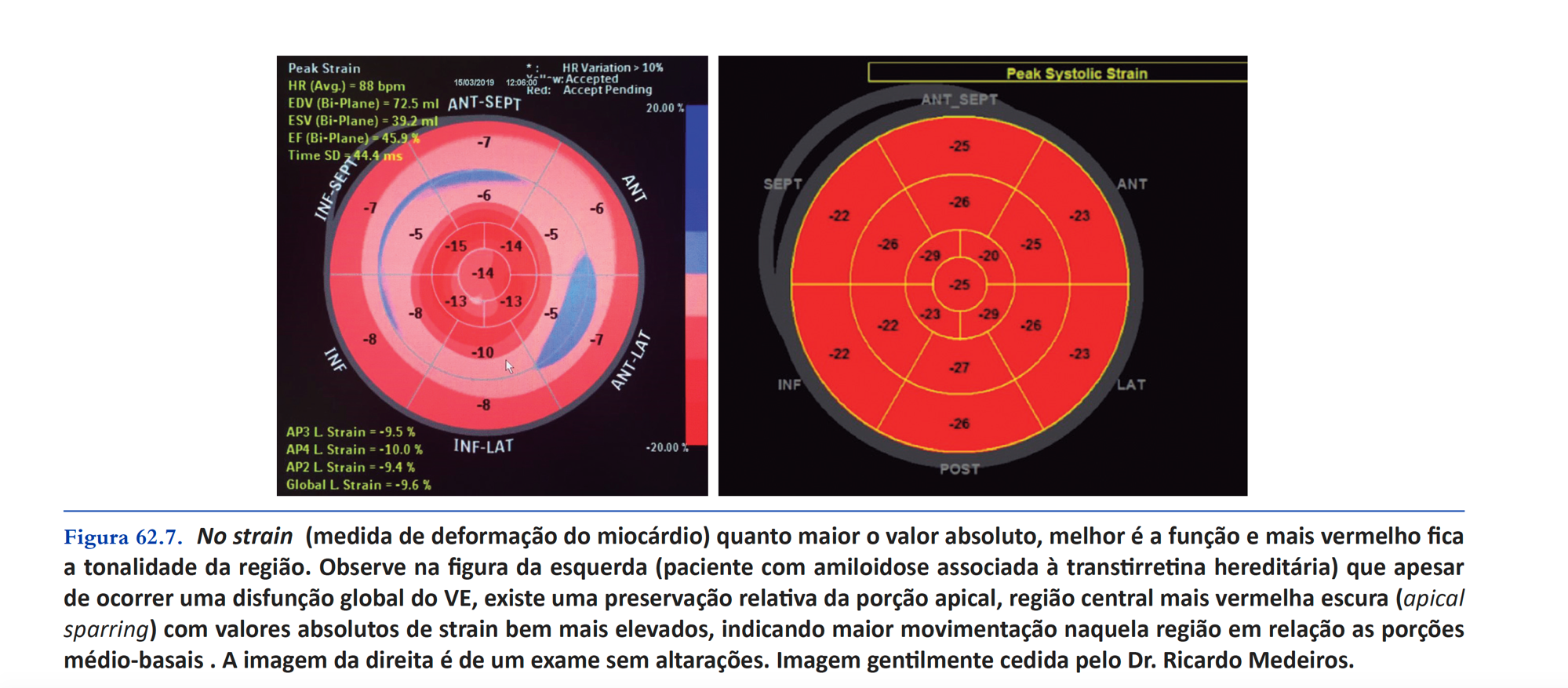
- Ecocardiograma se puede observar grosor ventricular y disfunción diastólica sin causa evidente o con estenosis aórtica que no justifica la gravedad de los hallazgos; como bajo flujo y bajo gradiente con FEVI preservado (paradójico); agrandamiento biatrial con ventrículos de tamaño normal ; aspecto cintilar hiperecogénico y heterogéneo “granulado” (granular sparkling) ; Strain longitudinal global reducida (SLG), estar por debajo del 12% se considera un marcador pronóstico (recordando que el valor normal es superior al 18% ), con preservación de la porción apical ( preservación apical o “signo de cereza en el pastel” ); ICFEP con disfunción del VD (turgencia yugular, hepatomegalia, ascitis). ! Cabe destacar que el 13% de los pacientes com ICFEP y pared del VI> 12 mm tienen amiloidosis cardíaca!
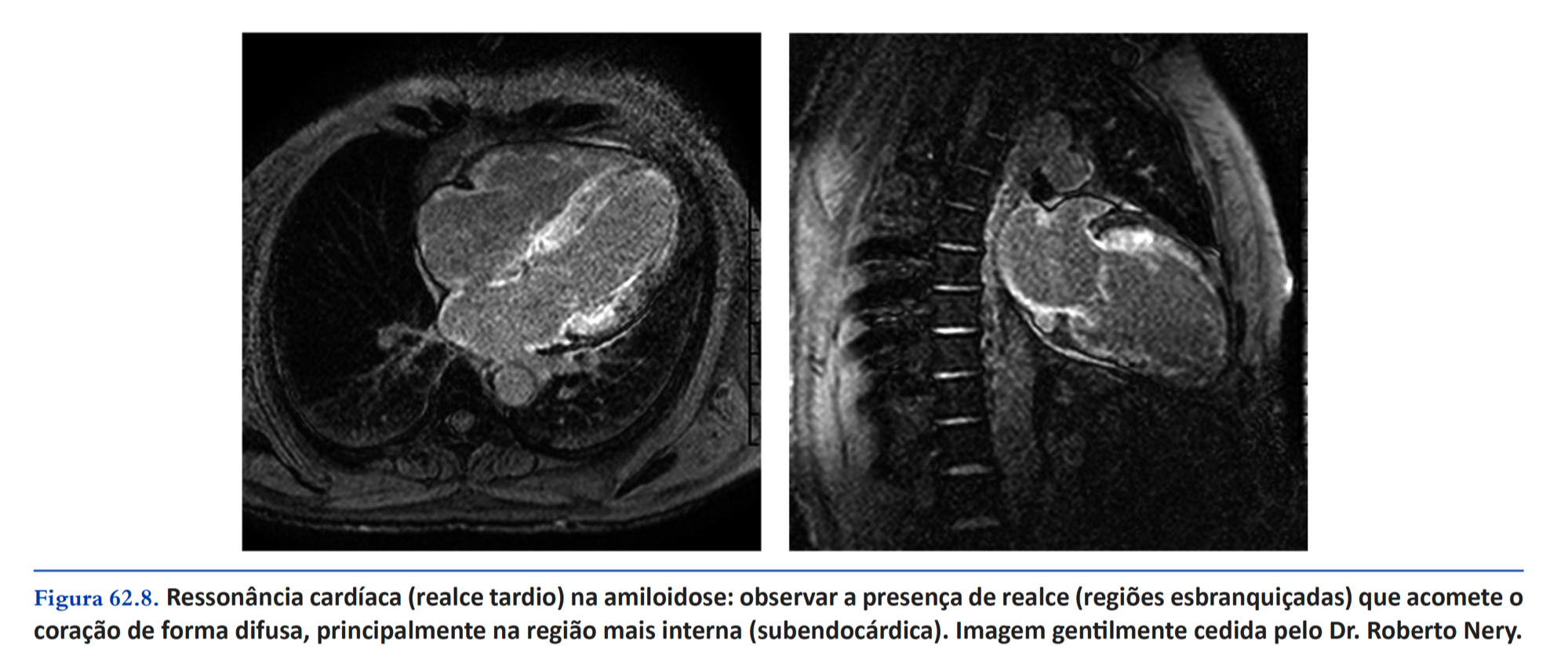
- Resonancia magnética cardíaca con realce tardio subendocárdico difuso y circunferencial del VI, con diferentes grados de extensión miocárdica, particularmente con un gradiente aumentado entre la base y el ápice. También aumenta la sospecha del hallazgo de T1 miocárdico nativo y volumen extracelular aumentado.
- Elevación desproporcionada de NT-proBNP en ausencia de insuficiencia cardíaca significativa y troponina elevada crónicamente en ausencia de síndrome coronario.
A continuación se muestra una imagen de nuestro libro Cardiología Cardiopapers 2da edición que muestra el “apical sparing”.
- Otra imagen de nuestro libro que muestra el realce tardio subendocárdica difuso em la RMC.
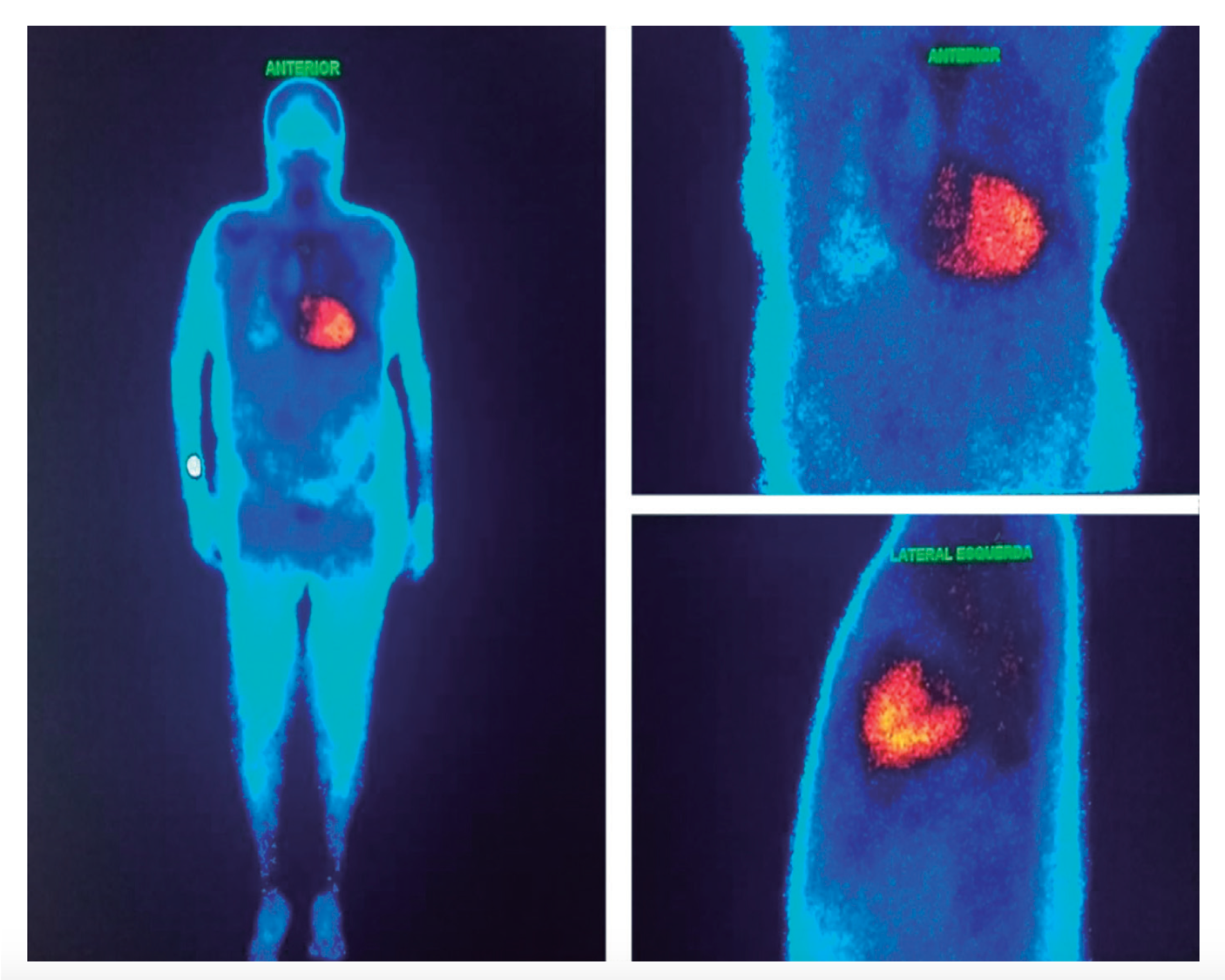
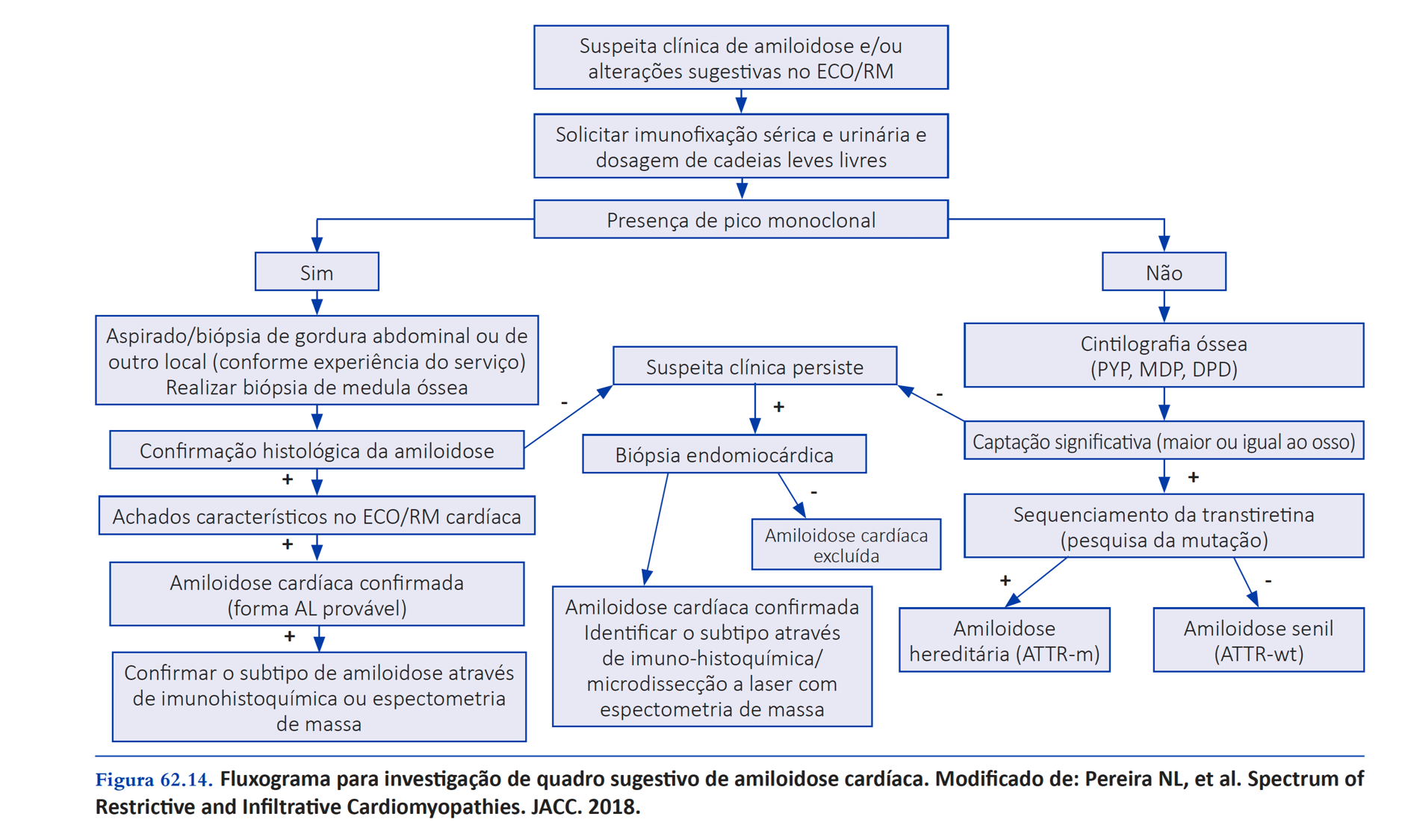
- Una gammagrafia ósea con difosfonato o pirofosfato de tecnecio-99mes muy útil en el diagnóstico diferencial de la amiloidosis cardíaca por TTR o AL porque tecnecio-99m se puede unir a las fibrillas de amiloide de TTR más intensamente que las fibrillas AL. Si la gammagrafía detecta la acumulación de TTR (captación grado ≥ 2), la investigación puede complementarse con un estudio genético para distinguir el tipo de TTR (mutante en TTR hereditario y normal en TTR salvaje). Los examenes indicados en la investigación de la amiloidosis AL son la electroforesis por inmunofijación de suero, orina y dosaje sérico de cadena ligera en busca de gammapatía monoclonal . La gammagrafía normal en ausencia de un pico monoclonal prácticamente impide el diagnóstico de amiloidosis cardíaca.
- Imagen de nuestro libro que muestra una gammagrafía miocárdica sugestiva de amiloidosis por transtiretina.
- Una biopsia extracardiaca positiva no es suficiente para confirmar el diagnóstico en pacientes sin hallazgos de imágenes típicos. Del mismo modo, una biopsia extracardiaca negativa no es suficiente para excluir el diagnóstico en pacientes con hallazgos de imagenes típicos.
- Los pacientes con estenosis aórtica y amiloidosis cardíaca coexistente son mas propensos de tener un patrón de flujo bajo y bajo gradiente (AVA <1 cm2, gradiente medio <40 mmHg e índice de volumen sistólico <35 ml/m2). La mitad de estos pacientes tienen la fracción de eyección preservada, es decir, tienen un patrón de estenosis aórtica paradójica. En estos casos, el eco de estrés com dobutamina a menudo no es concluyente y la tomografía con score de calcio se ha considerado la modalidad de imagen más adecuada para confirmar la gravedad de la estenosis aórtica.
- Debido al patrón restrictivo del llenado ventricular, el gasto cardíaco depende de la frecuencia cardíaca y, por lo tanto, los betabloqueantes deben usarse con precaución o evitarse. Los antagonistas de la renina-angiotensina pueden agravar la hipotensión sintomática. La digoxina se acumula en las fibrillas amiloides. Los diuréticos son esenciales para reducir la sobrecarga de volumen, pero también deben usarse con precaución. Debido al aumento del riesgo de trastornos graves de la conducción, el implante de marcapasos puede ser considerado para BAV primer grado, síncope de origen desconocido y distúrbios avanzados de conducción. El CDI para la profilaxis primaria sigue siendo controvertido. La anticoagulación sistémica no debe recomendarse de forma rutinaria, pero debe considerarse en pacientes con arritmias supraventriculares, eventos tromboembólicos previos y trombo cavitario.
- El tratamiento específico de la amiloidosis cardiaca implica la reducción de las proteínas amiloides:
- La amiloidosis AL se trata con quimioterapia (que tiene un alto riesgo de descompensar la insuficiencia cardíaca) y trasplante autólogo de células madre. En pacientes jóvenes con insuficiencia cardíaca sin otras comorbilidades, también se puede considerar la indicación temprana de trasplante de corazón.
- Para la amiloidosis TTR, existen numerosas estrategias farmacológicas en estudio, que incluyen: 1) silenciamiento de ARN TTR; 2) estabilización de TTR; 3) extracción / ruptura de fibrilla amiloide. El patisirany inotersen son silenciamiento de ARN de TTR estudió respectivamente en los ensayos Apolo y neuro-TT y fueron aprobados por la FDA para la enfermedad de neuropatía, pero no para cardiopatia (a pesar de que ambas han mostrado algún beneficio resulatdos en el corazón). Tafamidis y diflunizal interactuar con los sitios de unión de TTR, reduciendo su disociación y, en consecuencia, la amiloidogénesis. El tafamidis es el mejor estudiado hasta la fecha y fue aprobada por la FDA en 2019 para el tratamiento de la amiloidosis cardíaca, debido a los resultados ATTR-ACT, discutidos aquí . El beneficio del medicamento antiinflamatorio no esteroideo diflunizal en la amiloidosis cardíaca todavía está restringido a estudios pequeños. El PRX004 es un anticuerpo monoclonal en investigación experimental para limpiar formas tóxicas de amiloide TTR.
- El trasplante de corazón puede ser una opción viable (combinada con el trasplante de hígado en algunos casos de TTR hereditaria) dependiendo de la edad y las comorbilidades del candidato. La indicación de trasplante de hígado para TTR hereditaria ha disminuido dramáticamente con la llegada de los estabilizadores de TTR y no está indicada para TTR salvaje.
- La evidencia disponible sugiere que TAVI es mejor que la cirugía de reemplazo valvular en pacientes con amiloidosis cardíaca y estenosis aórtica, considerando que estos pacientes generalmente tienen un alto riesgo quirúrgico.
Resumen de cómo investigar a un paciente con sospecha de amiloidosis (diagrama de flujo de nuestro libro Cardiología Cardiopapers 2da edición):
Referencia: