Esta publicação também está disponível em:
 Português
Português
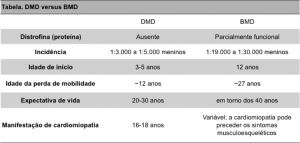
Distrofinopatias são um grupo de doenças neuromusculares distintas que resultam de mutações no gene da distrofina, com consequente ruptura do sarcolema e necrose na fibra muscular. São doenças de herança recessiva ligada ao cromossomo X e herdadas da mãe em 2/3 dos casos. Essas mães são em geral assintomáticas ou apresentam uma forma clínica branda, e tem um risco de 50% de terem outros filhos de sexo masculino afetados. No 1/3 restante dos casos, as mutações são espontâneas e o risco de recorrência para futuros filhos é desprezível. São conhecidos diversos tipos de distrofinopatias, mas a forma mais comum e letal é a distrofia muscular de Duchenne (DMD) que afeta cerca de um em cada 3-5 mil meninos. Outra forma mais rara é a distrofia muscular de Becker (DMB), caracterizada pelo início mais tardio e evolução menos severa (tabela).
A fraqueza muscular começa pelas pernas e compromete mais as cinturas escapular e pélvica do que pés e mãos. O achado laboratorial mais comum é a elevação da creatinoquinase (CK), de 10 a 100 vezes o valor de referência. O diagnóstico é feito através da biópsia muscular demonstrando a ausência de distrofina pelas técnicas de Western blot e imuno-histoquímica ou através de testes genéticos. A histologia é inespecífica e caracteriza-se pela substituição da fibra muscular por tecido conjuntivo, fibrose ou gordura.
As principais causas de morte são decorrentes do envolvimento muscular cardiorrespiratório. Estima-se que a incidência de cardiomiopatia1 na DMD aumente de 25%, aos 6 anos de idade, até mais de 90% nos pacientes que atingem a maioridade. No entanto, a restrição de mobilidade pode atrasar o diagnóstico da insuficiência cardíaca (IC). Na DMB a cardiomiopatia com CK elevada pode ser a única manifestação da doença, sem acometimento da musculatura esquelética associada. Recomenda-se o screening cardiológico no diagnóstico da distrofinopatia, sendo repetido a cada 02 anos até os 10 anos de idade e anualmente a partir de então. O padrão clássico de ECG apresenta ondas R proeminentes em derivações precordiais direitas, ondas Q profundas em precordiais esquerdas, intervalo PR curto e desvio de eixo para direita ou BRD. Taquicardia sinusal é a arritmia mais frequente, mas fibrilação/flutter atrial, taquicardia ventricular (TV) e BAVT foram descritos. Os achados de ECG costumam preceder as alterações do ecocardiograma. Os principais fatores de risco para TV são a fração de ejeção inferior à 35% e o QT longo. O ecocardiograma pode ser limitado pela pobre janela acústica associada à escoliose acentuada. Pode haver dilatação de câmaras, disfunção sistólica e/ou diastólica, alterações contráteis segmentares (principalmente na região posterior), trombos cavitários, disfunções valvares e derrame pericárdico. A ressonância magnética cardíaca (RMC) pode demonstrar realce tardio miocárdico inferior e posterior, consistente com os achados histológicos de fibrose, além de dimensionar a função ventricular e a dilatação de câmaras. O uso de biomarcadores como BNP e/ou troponina não está estabelecido para o screening de DMD.
Não existe cura para as distrofinopatias, mas a sobrevida aumentou com o suporte fisioterápico, nutricional, respiratório e ortopédico e o tratamento direcionado para IC. Corticóides melhoram a força muscular e parecem atrasar o início e a progressão da insuficiência cardíaca. Os esquemas propostos são prednisona 0,75mg/Kg/dia ou deflazacorte 0,9mg/Kg/dia. O uso de diuréticos e, principalmente, a modulação neuro-hormonal com iECA/BRA, beta-bloqueador e antagonistas do receptor mineralocorticóide, seguem a mesma orientação das diretrizes clínicas para IC com fração de ejeção reduzida e só devem ser prescritos nesses casos. Entretanto, existe evidencia de que a prescrição de iECA (perindopril) precoce possa atrasar o surgimento da IC. O transplante cardíaco é o tratamento definitivo para a IC terminal, embora o envolvimento respiratório e a dificuldade na reabilitação pós-operatória possam ser contraindicações relativas. Quando o transplante é bem indicado, a sobrevida livre de rejeições, infecções ou vasculopatia do enxerto é igual à de pacientes sem distrofinopatias. Os dispositivos de assistência ventricular (DAV) vem ganhando espaço como terapia de destino em portadores de DMD contraindicados ao transplante de coração. CDI não é indicado na prevenção primária de TV. Terapias de reparação da distrofina estão sob investigação.
Famílias com histórico de distrofinopatias devem procurar um serviço de aconselhamento genético que ofereça testes e consultas com médicos e geneticistas para diagnosticar, informar e identificar os riscos para os filhos.
Referência: Kamdar F, Garry DJ. Dystrophin-Deficient Cardiomyopathy. J Am Coll Cardiol 2016;67(21):2533-46.